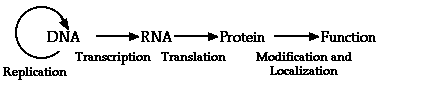
ISMB Tutorial 2
Bioinformatics and Molecular Biology
Douglas L. Brutlag
Stanford University School of Medicine
Stanford, California 94305-5307
(Much of the following material was extracted from Stanford’s course on Advanced Molecular Biology,
http://cmgm.stanford.edu/biochem201, and parts were authored by others. In particular, the section on transcription was written by by Roger Kornberg.).Abstract
This tutorial will present an overview of molecular biology; the problems it addresses and the methods it uses. We will present the flow of genetic information from the genome to the phenotype (biological characteristics) of an organism. We will present an overview of the molecular steps that nature uses to replicate and repair DNA; regulate expression of genes; process and translate messenger RNAs; modify and target proteins in the cell; integrate and regulate metabolism. We will describe, and give specific examples, where molecular processes can be addressed computationally. We will emphasize the need for multiple representations of many steps (heuristic, rule-based, probabilistic, information based, graph theoretic etc.). We will present a view of molecular biology as an information science. We will parallel the molecular and bioinformatic approaches to biological questions and describe the synergisms that result from using both.
Introduction
The molecular biological approach to understanding the flow and expression of genetic information involves studying the structure of macromolecules (DNA, RNA and protein) and the metabolic steps that mediate the flow of information from the genome to the phenotype of the organism. With the advent of large databases of genetic information for these macromolecules, a completely new approach to studying gene expression and its regulation has developed. This field is known as bioinformatics.
Bioinformatics is a subset of a larger area of computational molecular biology which includes the application of computer and information science to areas such as genomics, mapping, sequencing and determination of sequence and structure by classical means. The primary goals of bioinformatics are predicting three-dimensional structure from primary sequence, predicting biological or biophysical functions from either sequence or structure, and simulating metabolism and other biological processes based on these functions. Many methods of computer and information science are applied to these goals including machine learning, information theory, statistics, graph theory, algorithms, artificial intelligence, stochastic methods, simulation, logic, etc. The purpose of this tutorial is to demonstrate how these methods can be used to discover biological first principles from data residing in the sequence and structure databases.
Molecular biologists study the flow of genetic information from the genome to phenotype of the organism. The simplified diagram below shows the textbook view of this information flow, including the informational molecules that most biologists study, DNA, RNA and protein and the processes that relate them, replication, transcription, translation and protein targeting.
It is the structure and function of the molecules shown above that is of critical importance to the flow of information both from genome to phenotype and also from generation to generation. The processes represented by the arrows (replication, transcription, translation, and protein targeting) mediate and control the expression of the genetic information. The goal of molecular biology is to understand the mechanism, specificity and regulation of these processes.
There is another view of the flow of genetic information that is more amenable to attack by bioinformatics:
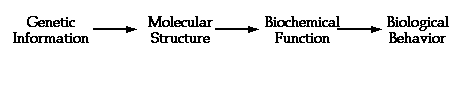
Each step in the pathway above poses a challenging problem for the information scientist or bioinformatician.
Molecular modeling and molecular dynamics are two of many methods used to predict structure from function . Several methods of machine learning are used to predict function from sequence or structure. There are also many methods for simulating metabolism given known biological functions .
Medical science would like to understand this pathway well enough to know which genomic changes could give rise to each known inherited disease. The pharmaceutical industry would like to use knowledge of this pathway to produce drugs, proteins or genetic therapies that can reverse disease phenotypes.
The international efforts to determine complete sequences of bacterial and eukaryotic genomes makes it imperative that we be able to interpret the genetic information directly. There are far more genes and proteins whose sequences are known than can be analyzed by classical biophysical, biochemical or biological methods. For example, in the last 100 years the worlds biochemists have purified and characterized only 12,000 enzymes to the point where we can understand their function and phenotype when inhibited or mutated. It has taken over 40 years for all the world’s biophysicists to determine a similar number, 10,000 three-dimensional structures. Whereas, it has only taken twenty years since the advent of cloning and sequencing methods to determine the complete genetic information of several viral and bacterial genomes and over 1,500,000 genes and their protein products. This rapid pace of sequence determination mandates that we be able to recognize structures and functions which we have seen before.
In order to leverage this valuable genetic information, we must learn to classify new protein sequences into existing superfamilies and new genes into existing phylogenies, so that we can commit valuable laboratory time to study the truly novel genes and proteins. More importantly, it is also critical that we be able to reason from examples and deduce novel structures and functions from sequence databases. The purpose of this tutorial is to review the major methods currently in use, their limitations and the leading areas of bioinformatics research.
The Challenge
Elucidating structure and function from sequence is a challenging problem for several reasons. First, genetic information is highly redundant. In coding regions, for example, each amino acid has on the average 3.05 codons to chose from (61 non-terminating codons/20 amino acids). This means that even a short protein like human insulin, 51 amino acids in length, has 351 or 1024 distinct DNA sequences that can encode an identical amino acid sequence.
There is structural redundancy in genetic information as well. There are over 700 globin sequences in the current protein databases. All of these proteins have a nearly identical three-dimensional structure, but the sequences are so different as to be unrecognizable by most methods currently in use. This implies that many different protein sequences can encode precisely the same folding, just as many different DNA sequences can encode the same protein sequence.
Nature makes extensive use of this redundancy to encode multiple functions in genetic information. Within a single eukaryotic coding region, the DNA sequence will show evidence for codon choice that regulates the rate of translation, dinucleotide selection that favors bending of the DNA into chromatin, as well as the protein coding information itself. Finally, the genetic information is inherently one-dimensional, but structure and function depend on three-dimensional attributes.
The Structure of DNA
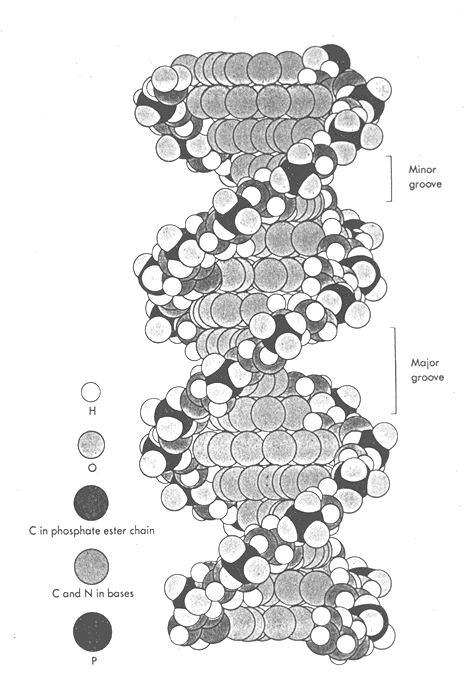
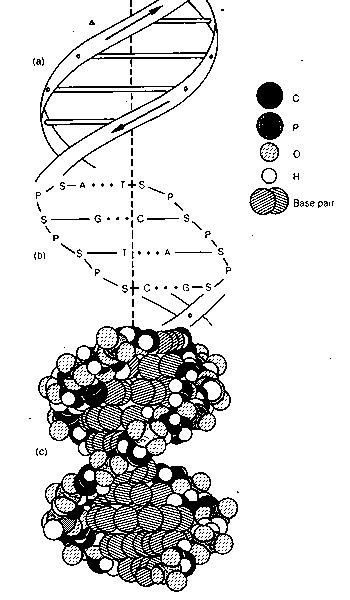
The structure of DNA is much more flexible than previously conceived. DNA is a highly flexible molecule that can undergo a series of transformations leading to many conformations with different biological functions.
2. The structure of DNA as originally proposed by Watson and Crick depended on one major assumption, that the structure of DNA was independent of its sequence.
3. We now know from X-ray crystal structure analysis of DNA segments, that the structure of DNA varies dramatically with sequence.
4. The Watson-Crick structure had many implications for the replication and transcription of the genetic information.
5. The variability of the structure of DNA also has critical implications for the regulation of expression of the genetic information.
A. The first important consequence of the Watson-Crick model of DNA was that the molecule was double helical and that the two strands contained complementary base sequences. This meant that the genetic information is redundant. This redundancy allows repair of damaged DNA and simplifies replication of the DNA via strand separation.
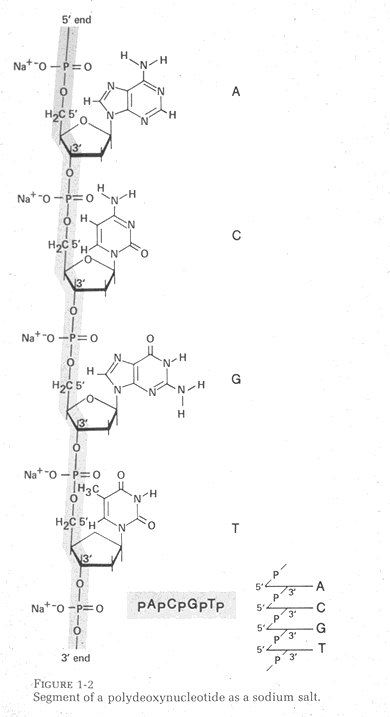
B. Another important consequence of the Watson-Crick structure was the anti-parallel nature of the DNA chains. Anti-parallel chains cause considerable difficulty for replication and transcription.
C. The similarity of the DNA base pairs to each other also makes faithful replication with less than one mistake in a hundred million a formidable task.
D. The structural similarity of diverse sequence makes recognition of genetic control sites difficult.
Watson-Crick B-Form DNA
1. A review of the origin and the experimental support for Watson and Crick's structure for the B-form of DNA.
2. From basic chemical analysis, Watson and Crick knew:
A. DNA contained long polymeric chains.
B. DNA contained deoxyribonucleosides in 5'Æ 3' phosphodiester linkage.
C. X-ray diffraction suggested a helical structure.
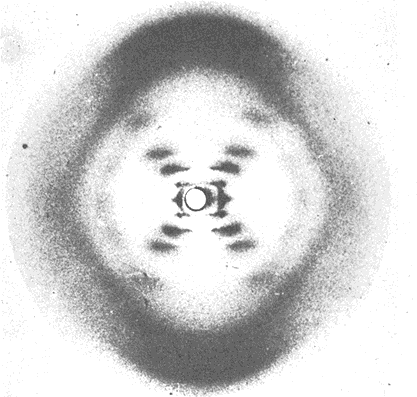
- The cross pattern suggested a helical pitch angle about 45°.
- Axial reflections gave repeating units of 3.4 and 34 Å.
- Radial reflections gave a fiber width of 20 Å.
D. Chargaff's work showed that the base composition of DNA varied from organism to organism but certain relationships between the amounts of various bases always held. These relationships are called Chargaff's rules:
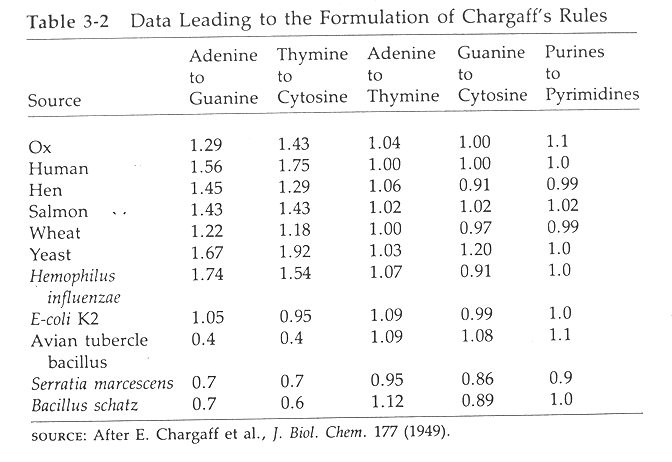
- The amount of adenine equals the amount of thymine.
- The amount of guanine equals the amount of cytidine.
- The amount of adenine plus guanine equals 50% of the total
implying that 50% of the bases in DNA are purines.
- The amount of thymine plus cytosine equals 50% of the total
implying that 50% bases in DNA are pyrimidines.
E. Watson and Crick proposed base pairing rules to explain Chargaff's equalities.
- They chose base pairs connected by hydrogen bonds.
- The bases were in their normal tautomeric forms (uncharged) at pH 7.0.
- They picked an AT pair and a GC pair that gave superimposable locations of the glycosylic linkages. A consequence of this is that the structure of the DNA would be sequence independent.
- The first base pairs observed in X-ray crystallography experiments were the Hoogstein base pairs and not the Watson and Crick base pairs.
F. The deoxyribose of each base pair are attached in opposite orientation.
G. Each base was in the anti-conformation.
H. This conformation of nucleosides resulted in the opposite polarity of DNA chains in the resulting helix.
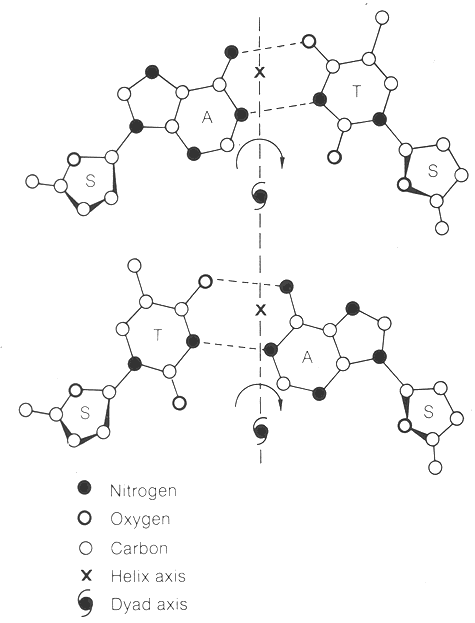
I. This conformation and resulting antiparallel chains generate an axis of dyad symmetry (axis of two-fold rotational symmetry) at each base pair. Dyad axes are very important for proteins that bind to DNA. Most DNA-binding proteins possess an axis of symmetry and bind to symmetric DNA sequences.
J. This structure has the base-pair as the primary repeating unit and results in an additional axis of symmetry between each base pair.
K. Watson and Crick then connected base pairs with phosphodiester bonds that spaced the bases 3.4 Å apart and rotated each subsequent base pair by 36°. This rotation generates a right-handed double helix with 10 base per turn and repeating elements every 3.4 and 34 Å.
L. The obtuse angle of the glycosylic linkages leads to major and minor grooves in helix with specific groups in each group.
M. The bases were perpendicular to the helix axis.
Crystal Structure of B-DNA
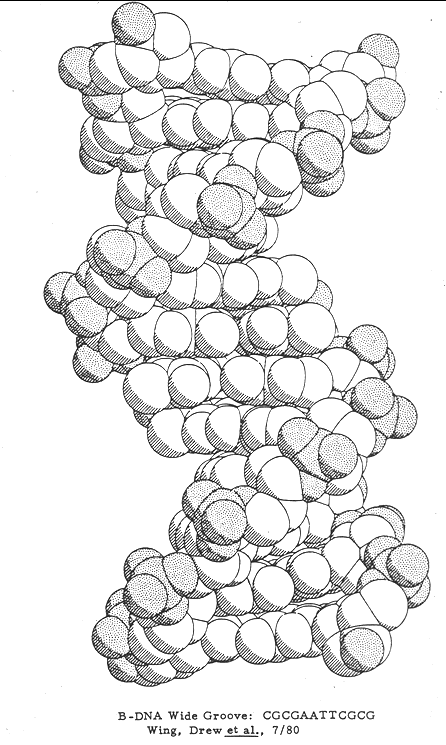
1. Thirty years after the Watson-Crick proposal, Dickerson and Rich determined the complete structure of crystalline DNAs. Advances in the synthesis of large quantities of short synthetic segments of DNA allowed each of them to crystallize a unique DNA sequence.
2. Dickerson's B form crystals confirmed in most part the Watson-Crick model. DNA was double helical with antiparallel strands. The bases associated in Watson and Crick base-pairs with hydrogen bonds in the center.
3. There were also two major differences.
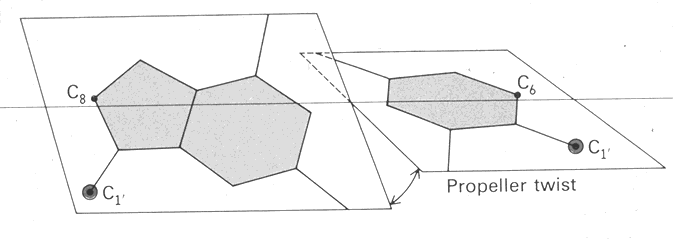
A. First, the base pairs were not flat, but were twisted with respect to each other. This was called a propeller twist.
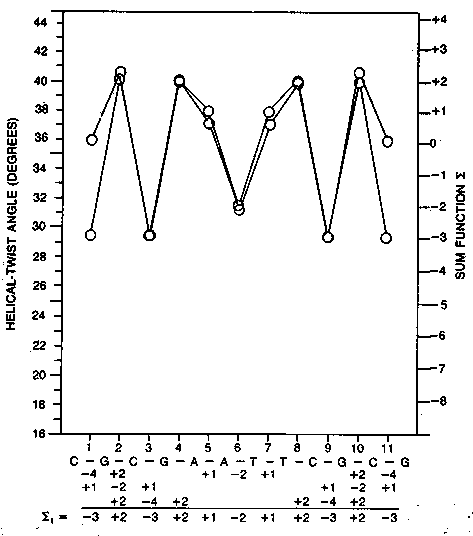
B. The rotation from one base pair to the next was not a constant 36° as predicted, but instead varied from 27° to 40°. This variation in twist angle was extremely important because it implied that the structure of B-DNA was sequence dependent.
4. The propeller twist of the base pairs results in purine-purine clash in the center of the helix. Because the purines are larger than the pyrimidine rings, they extend beyond the helical axis of DNA.
5. DNA attempts to reduce purine-purine clash in several ways:
A. The base pairs rotate less along the helix axis in the purine-pyrimidine sequences (lower average helical twist). They tend to rotate less in the pyrimidine-purine sequences (lower than average helical twist). The average helical twist was still very close to the 36° proposed by Watson and Crick.
B. Another way DNA minimizes the purine-purine clash is that it bends toward the minor grove or major groove to reduce the interaction.
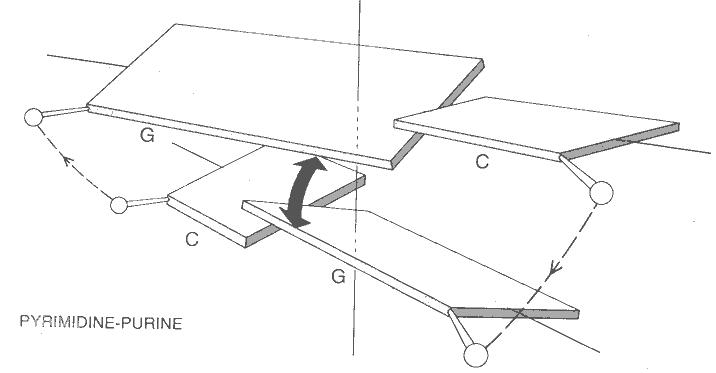
C. Finally clashing base pairs could slide left or right toward the phosphodiester backbones to minimize the purine-purine interaction.
6. The most important implication of these structural variations is that the actual structure of DNA depends strongly on the sequence. The positions of the phosphate groups, the positions of the amino and keto groups in DNA reflect the sequence in a predictable way. Current research is aimed at understanding this structural code and to determine if regulatory sequence-specific DNA binding proteins make use of this variation in recognizing DNA.
A-form of DNA
1. There are also many other forms of DNA more distinct from the B form that are biologically important. Most well known is the A-form which DNA assumes during dehydration or in RNA-DNA hybrid helices.
2. The base-pairs are not perpendicular to the helical axis but instead they are tilted at a steep angle.
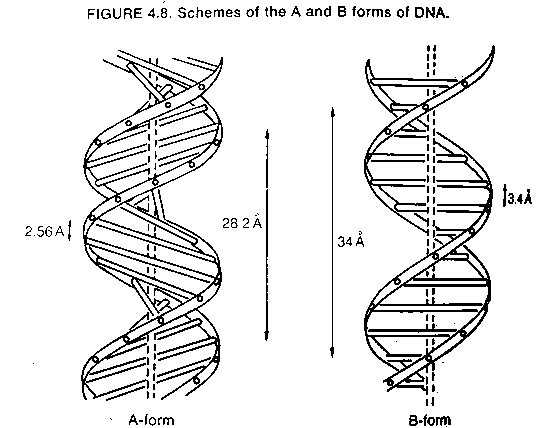
3. In the A form, the base pairs are also closer together along the helical axis; 2.55 Å center-to-center distance.
4. The helical pitch of A-form DNA is closer to 11 base pairs per turn in 28 Å rather that 34 Å. As a result, the A-form is 25% shorter than the B-form. DNA shrinks when it dries.
5. If binding of protein to DNA removes water it may result in altered conformation of the DNA thus stabilizing the interaction.
6. The tilted base pairs allow room for the 2' oxygen present in RNA chains and therefore all double helices containing at least one RNA strand are present in the A-form.
7. Duplex RNA (such as found in the replication intermediates of many viral RNAs such as polio virus RNA) is always in the A-form.
Z-form of DNA
1. When the self-complementary polymer (CG)3 was crystallized in high ionic strength conditions, a very unusual form of DNA called the Z-form was discovered.
2. The Z-form differs from the B-form in several ways:
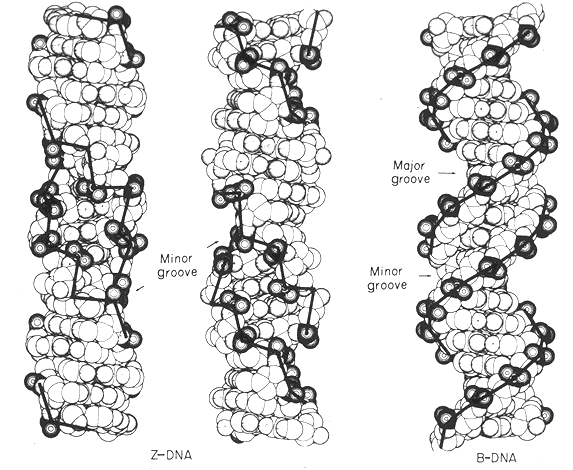
A. The helix was left-handed instead of right-handed.
B. The helix showed only a single groove rather than two.
C. The nucleotides along one strand alternate between the syn- and anti-conformation. The guanosines are all in the syn conformation while the cytidines are all in the anti conformation like the B-form.
3. In most other respects the two forms are similar:
A. Both forms are double helical and both have two chains of opposite chemical polarity.
B. Watson and Crick hydrogen bonds hold the chains together.
4. Since conformations between the purine and the pyrimidines in Z-DNA alternate, the basic repeating unit is no longer the base pair, but is a dinucleotide. This implies that there is no axis dyad symmetry at each base pair, only between base pairs.

5. Even the best evidence for Z-DNA in nature is controversial. Some of the best evidence comes from experiments involving antibodies directed specifically at the Z-DNA structure. Some authors have demonstrated that the presence of the Z-form of the DNA in these cytological preparations is an artifact of the preparation and if one prepares chromosomes carefully, no or very little Z-DNA antibody will bind.
6. The most one can say is that Z-DNA can form under physiological conditions in natural DNA sequences in which purines alternate with pyrimidines. Whether Z-DNA does form in cells and whether nature takes advantage of this unusual form is still speculation.
7. The possibility that DNA can assume two structures as distinct as the Z-form and the B-form shows that the chains are capable of much more flexibility than many had considered possible before.
Novel DNA Structures
1. Several novel forms of DNA involving the pairing of more than two strands and also forms involving parallel chains have been described. These structures generally form with specific DNA sequences and may have profound biological consequences.
A. Wells and others have evidence showing that oligopurine-oligopyrimidine sequences can fold back on themselves to form an internal region containing one triple stranded region and one single-stranded region. The third strand is base-paired in the major groove of a normal DNA duplex using hydrogen bonds similar to those found in the Hoogstein base pairs.
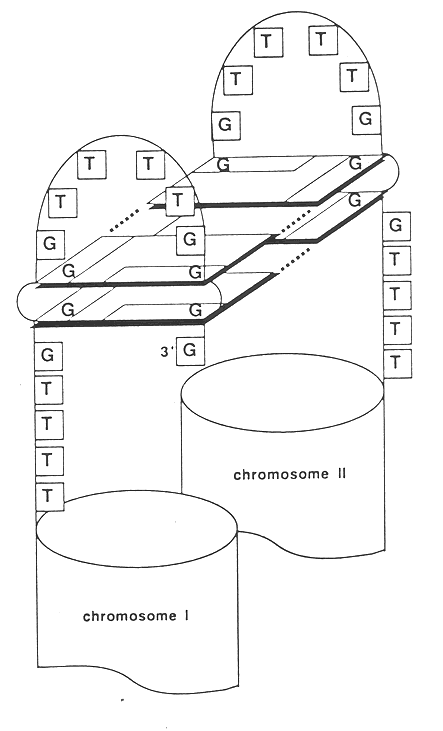
B. Sen and Gilbert have reported that DNA helices containing specific guanine rich sequences can self-associate to form four-stranded structures. The two helices are hydrogen bonded together by Hoogstein pairing.
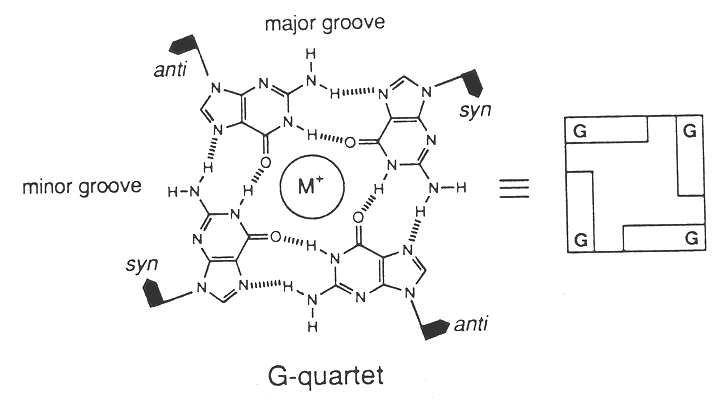
C. Tom Cech and Aaron Klug’s laboratories have demonstrated that sequences found at the ends of eukaryotic chromosomes can also form specific tetranucleotide base pairs, referred to as G-quartets that may be involved in holding chromosome ends together during mitosis.
D. Englund and others noticed that certain DNA sequences had an unusual migration during electrophoresis. Analyses of such DNAs have shown that they often contain runs of 3-4 As or Ts in a row and that these runs are repeated every 10 base pairs. Such runs result in bending of the DNA towards the minor groove and the repeating nature makes the DNA helix as a whole bend in one direction.
E. Tom Jovin and Johan van de Sande have demonstrated that specific AT rich DNA sequences can base pair to form a parallel double-helical structure. These structures are physically very similar to the B-form of DNA but they are ineffective as substrates for many enzymes.
References
Dickerson, R. E. (1992). DNA structure from A to Z. Methods Enzymology 211, 67-111.
Hagerman, P. J. (1990). Sequence-Directed Curvature of DNA Annu. Rev. Biochemistry 59, 755-781.
Herbert, A., & Rich, A. (1996). The biology of left-handed Z-DNA. J Biol Chem, 271(20), 11595-8.
Johnston, B. H. (1992). Generation and detection of Z-DNA. Methods Enzymol 211, 127-58.
Rich, A., Nordheim, A. and Wang, A. H. J. (1984). The Chemistry and Biology of Left-Handed Z-DNA. Annual Review of Biochemistry, 53, 791-846.
Rich, A. (1993). DNA comes in many forms. Gene, 135(1-2), 99-109.
Rich, A. (1994). Speculation on the biological roles of left-handed Z-DNA. Ann N Y Acad Sci, 726, 1-16; discussion 16-7.
Rippe, K. and Jovin, T. M. (1992). Parallel-stranded duplex DNA. Methods Enzymol 211, 199-220.
Travers, A. A. (1990). Why bend DNA? Cell, 60(2), 177-80.
Watson, J. D. and Crick, F. H. C. (1953a). Molecular structure of nucleic acids. Nature 171, 737-738.
Watson, J. D. and Crick, F. H. C. (1953b). The structure of DNA. Cold Spring Harbor Symp. Quant. Biol. 18, 123-131.
Wells, R. D. (1988). Minireview: Unusual DNA structures. J. Biol. Chem. 263, 1095-1098.
Williamson, J. R. (1994). G-quartet structures in telomeric DNA. Annu Rev Biophys Biomol Struct, 23, 703-30.
Internet Resources
Structure Database
Molecules R US http://molbio.info.nih.gov/cgi-bin/pdb
RasMol Distribution
http://www.umass.edu/microbio/rasmol/Chime Plugin
http://www.umass.edu/microbio/chime/
Kinemage Program
http://www.faseb.org/protein/kinemages/MageSoftware.htmlSwiss-Prot Viewer
http://www.expasy.ch/spdbv/mainpage.htmProtein/DNA Kinemages
http://www.faseb.org/protein/ProTeach/
RNA Structure
Introduction
 Unlike DNA, whose primary role is to store the genetic information, RNA plays many roles in the cell. RNA is structurally much more flexible than DNA because it is usually a mixture of single-stranded and duplex regions rather than predominantly duplex. The presence of the 2’-hydroxyl chemical group also makes RNA chemically more labile and not generally suitable as a molecule for storage of large amounts of genetic information over long periods of time. RNA is readily cleaved in alkaline conditions and in the presence of divalent metal ions, both of which deprotonate the 2’ hydroxyl group, which then attacks the phosphate esterified at the 3’ position, resulting in chain scission and a 2’,3’ cyclic phosphodiester terminal group.
Unlike DNA, whose primary role is to store the genetic information, RNA plays many roles in the cell. RNA is structurally much more flexible than DNA because it is usually a mixture of single-stranded and duplex regions rather than predominantly duplex. The presence of the 2’-hydroxyl chemical group also makes RNA chemically more labile and not generally suitable as a molecule for storage of large amounts of genetic information over long periods of time. RNA is readily cleaved in alkaline conditions and in the presence of divalent metal ions, both of which deprotonate the 2’ hydroxyl group, which then attacks the phosphate esterified at the 3’ position, resulting in chain scission and a 2’,3’ cyclic phosphodiester terminal group.
 Single stranded regions of RNA are also very susceptible to attack by ribonucleases in the cell. Many RNAs, especially messenger RNAs, have a lifetime less than one cell division. This short lifetime is essential for cells to respond to changing environmental conditions. Other RNAs, such as ribosomal RNAs, tRNAs and small nuclear RNAs have considerable secondary structure and some are protected by complexes with proteins, stabilizing them further.
Single stranded regions of RNA are also very susceptible to attack by ribonucleases in the cell. Many RNAs, especially messenger RNAs, have a lifetime less than one cell division. This short lifetime is essential for cells to respond to changing environmental conditions. Other RNAs, such as ribosomal RNAs, tRNAs and small nuclear RNAs have considerable secondary structure and some are protected by complexes with proteins, stabilizing them further.
In the past 10 years, it has been reported that many RNAs can even catalyze enzymatic reactions. Such RNAs are known as ribozymes and included among them are self-splicing Group I introns, hairpin ribozymes, and hammer head ribozymes. Most recently, ribosomal RNA itself has been shown to play an essential role in peptidyl transfer. RNA and RNA protein complexes can mediate several reactions suggesting that the original catalysts in the evolution of life may have been RNA rather than proteins (the RNA world hypothesis).
Understanding the structure of these RNAs is critical to understanding their function including how they can catalyze chemical reactions with the requisite specificity and acceleration. For the past 15 years only a couple of RNA structures had been determined because of the difficulty of crystallizing such flexible and sensitive molecules. By changing the ribozyme structure so that is no longer acts on its substrate, or by using solution methods for determining their structure (NMR and fluorescent energy transfer) a number of novel ribozyme structures have been determined with sufficient accuracy to allow speculation as to their mechanism. In this section we will discuss the structure of RNA and how that structure leads to its function.
Base pairing and stacking in determining RNA Structures
RNA structure largely derives from base pairing and base stacking. In addition to the standard Watson-Crick base pairs, there are numerous other parings of bases that can stabilize RNA tertiary structures including G-A mismatched pairs (Hoogstein-like base pairs), triple base pairs and G-U base pairs. The latter base pair does not destabilize RNA stems and can add significant stacking energy by permitting a stem to continue. Initially it was suggested that base pairing was responsible for the stability of RNA stems and duplexes. It was quickly discovered that it was the stacking of adjacent base pairs which was more important in determining the stabilization energy of stems and helices. However, there are several instances in tRNA and other structures where individual base pairs can indeed form and have a stabilizing effect on the structure as a whole.
In addition to the stabilizing effect of base pairs, there are numerous destabilizing structures within RNAs. These include single base mismatches, interior loops of more than one mismatch, bulge loops in which many bases are single stranded, three and four way branches, single-stranded hairpin loops, etc. While energies have been calculated for base-pairs from model systems, the destabilization energy of these mismatched regions has only been estimated.
Much effort has been expended attempting to predict the folding of RNA molecules from the sequence alone. The methods attempted include:
1) Maximizing base pairs (poor)
2) Maximizing the base pair energies (incorrect)
3) Maximizing stacking energy (better)
4) Maximizing the total energy (best)
One of the major problems with all of these methods is that they do not take into account the fact that RNA is flexible and it can form many competing structures. It is critical to evaluate all of these competing structures in the same was as they would form during RNA folding in the cell. If, like in the case of protein folding, there are proteins that catalyze the RNA folding process, it may not be possible to predict the structure most stabilized. However, one of the most successful RNA folding programs by H. Martinez, does take into account many of the possible structures and actually proposes a large number of potential structures all within a specified number kilocalories of the most stabile form. Martinez’s paper on an RNA folding rule describes an algorithm which folds RNA as one might expect it occurs in vivo, folding the most stable secondary structure element first, then the next, then the next etc.
One of the best examples of competitive RNA structures in vivo is in the regulation of the tryptophan operon by attenuation.
Another important aspect of RNA folding is the topology of the folding. If two overlapping regions of RNA can pair with another region, the RNA can become locked in a structure known as a pseudoknot. Most RNA structure prediction programs are not capable of detecting or prediction regions capable of forming pseudoknots.
tRNA structures
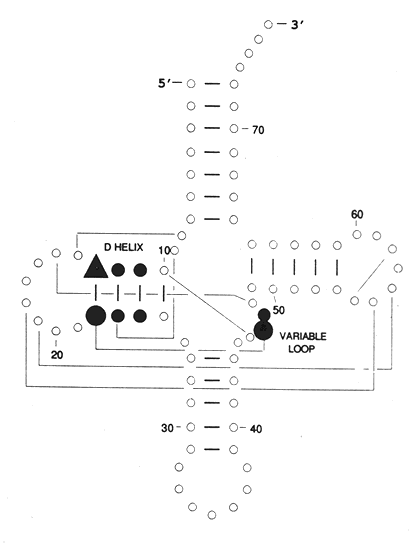
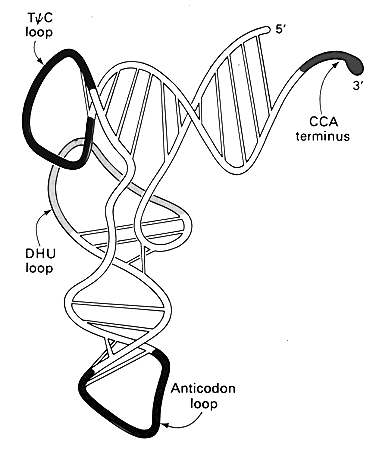
tRNA structures illustrate each of the points made in the previous section. The very first tRNA sequence was determined by Holley and his coworkers, for which they received the Nobel Prize. The sequence itself immediately suggested a cloverleaf secondary structure because of the presence of four long self-complementary sequences. This cloverleaf structure included four stem loops with a central four-way branch. However, it was not until the determination of the actual structure of tRNA by crystal structure methods, was the L-shape structure appreciated. Two pairs of stems stacked on top of each other and several bases in two of the loops paired with each other holding the entire structure into an L shape with an angle estimated to be between 80 and 130°.
RNA structures and base pairings are now often predicted from phylogenetic sets of RNAs from many organisms. By looking for positions that are always correlated with each other, one can deduce base pairing in the absence of physical evidence. One can also predict stacking interactions and even interactions with other molecules such as the tRNA synthases.
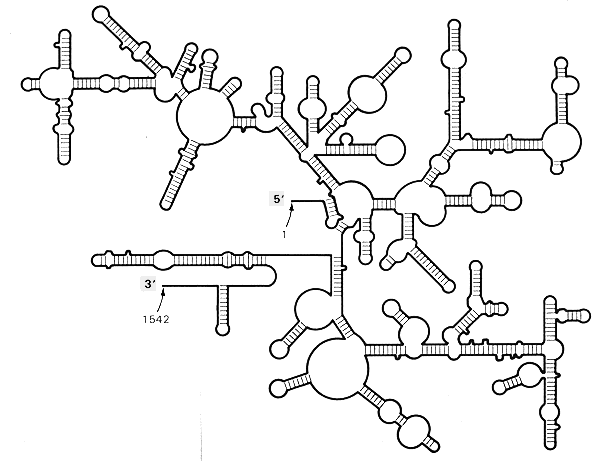
Ribosomal RNAs
The 16 and 23 S ribosomal RNAs are exceptionally long and difficult structures to determine. Some of the methods used, besides computer search, include chemical and nuclease sensitivity, cross-linking agents, protein-RNA cross-links, fluorescent transfer, 2 dimensional NMR etc. A picture of the current state of knowledge of the potential structural elements in these large RNAs is kept on the RiboWeb page on the Internet. The ribosomal RNAs also interact with the many ribosomal RNA proteins, further stabilizing certain of the base interactions.
Hammerhead Ribozymes
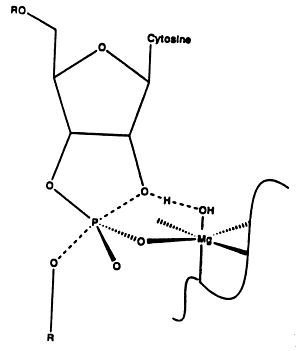
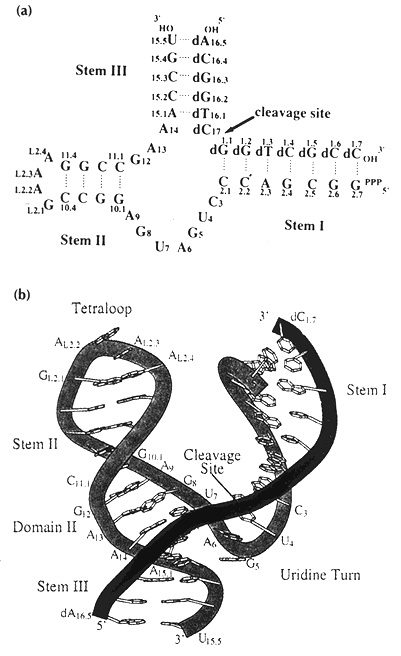 Hammerhead RNAs are small self-cleaving RNAs that contain a conserved motif found in several viroids and viral satellite RNAs that are self replicating via a rolling circle mechanism. The structure involves three stems, usually labeled I, II and III, with stem III being a short (5 base pair) stem nearly collinear with stem II. Stem I forms a three way junction with stems II and III and stem I contains the C-17 residue which contains the scissile bond cleaved the ribozyme. There are 15 highly conserved residues in the neighborhood of this bond, nine of which are not base paired. A run of four purines (GAAA) is in close apposition to the cytosine-17 where the cleavage occurs. Crystal structures indicate the presence of a divalent metal binding site in this region as well. Many catalytic ribozymes contain a stem loop with the sequence GARA (where R can be either A or G) in the loop being involved the catalytic site. The stacking of these purines is believed to form a rigid structure that can coordinate a divalent metal ion that can serve to catalyze strand cleavage.
Hammerhead RNAs are small self-cleaving RNAs that contain a conserved motif found in several viroids and viral satellite RNAs that are self replicating via a rolling circle mechanism. The structure involves three stems, usually labeled I, II and III, with stem III being a short (5 base pair) stem nearly collinear with stem II. Stem I forms a three way junction with stems II and III and stem I contains the C-17 residue which contains the scissile bond cleaved the ribozyme. There are 15 highly conserved residues in the neighborhood of this bond, nine of which are not base paired. A run of four purines (GAAA) is in close apposition to the cytosine-17 where the cleavage occurs. Crystal structures indicate the presence of a divalent metal binding site in this region as well. Many catalytic ribozymes contain a stem loop with the sequence GARA (where R can be either A or G) in the loop being involved the catalytic site. The stacking of these purines is believed to form a rigid structure that can coordinate a divalent metal ion that can serve to catalyze strand cleavage.
The structure of the hammerhead ribozyme determined by X-rays and that by fluorescent transfer show some differences. First, the crystal structure of an all-RNA ribozyme was synthesized with a GUAA sequence in place of the highly conserved GAAA sequence. Secondly, the catalytic reaction is blocked by a 2’O methylation of the active site cytosine residue. The structure also contains two Hoogstein GA base pairs near the active site.
Protein Structure
Introduction
Proteins are more flexible than nucleic acids in structure because of both the larger number of types of residues and the increased flexibility and lower charge density of the polypeptide backbone. Proteins can serve many roles in the cell; as enzymes, as structural components, membrane components, as templates, as substrates and as products of reactions. Many aspects of protein metabolism are catalyzed and regulated by the cell. These include their rates of expression, their translation, their folding, their targeting to the proper cellular location and their degradation. Proteins, and the functions they catalyze, are the end-product of the genes that encode them. One of the most important functions of proteins is to regulate the expression of other proteins.
In this section we will discuss the components of proteins, their covalent structure, their non-covalent interactions, higher order structures such as motifs and domains and then give several examples of different types of protein folds. It will be extremely useful for you to down load the Kinemage 4.2 program and the Proteach Kinemage collection for reviewing the material presented in the tutorial. Pointers to the locations to obtain this program and the Proteach files are on the course Web page.
Amino Acids
The amino acid residues of proteins are defined by an amino group and a carboxyl group connected to an alpha carbon to which is attached a hydrogen and a side chain group R. The smallest amino acid, glycine, has a hydrogen atom in place of a side chain. All other amino acids have distinctive R groups. Because the alpha carbon of the other amino acids have four different constituents, the alpha carbon atom is an asymmetric center and most naturally occurring amino acids are in the L form.
Amino acids fall into several naturally occurring groups including hydrophobic. hydrophilic, charged, basic, acidic, polar but uncharged, small polar, small hydrophobic, large hydrophobic, aromatic, beta-branched, sulfur containing etc. Hydrophobic amino acids are sometimes called non-polar and reside primarily on the interior of the protein. Hydrophilic amino acids are sometimes called polar and reside primarily on the exterior of the protein. Many amino acids will fall into more than one group since each amino acid side chain has several properties.
Peptide Bonds
Amino acids are linked to each other by peptide bonds. Peptide bonds are formed by the dehydration of the carboxyl group of one amino acid and the amino group of the next. Because of the resonance structure of the electron orbitals on the amino and carboxyl groups, the peptide bond is planar.
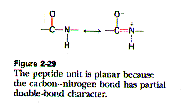
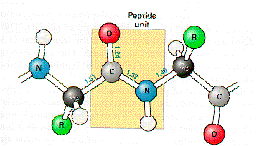
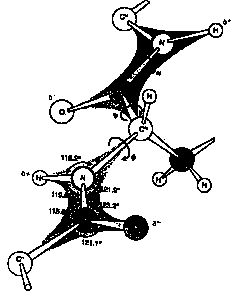
The dihedral angle between the amino group and the alpha carbon and the alpha carbon and the carboxyl group are free to rotate and these angles are referred to as the phi-psi angles.
Glycine, with the smallest side chain, has the most conformational flexibility about the phi-psi angles. Other amino acids are restricted in their rotation due to steric hindrance from the side chains. The rotation of the dihedral angles of side chains about the different bonds, referred to as chi-1, chi-2 etc., are also restricted for different side chain elements. Proline, which in which the side chain is linked back to the backbone is the most restricted, Only two conformations are permitted.
Forces determining protein structure
There are several covalent and non-covalent forces that determine protein structure. The list of forces include (not exhaustive):
1) van der Waals interactions between immediately adjacent atoms. These non-covalent forces result from the attraction of one atoms nucleus for the electrons of another atom in a non-covalent form (no sharing of orbitals). These forces are much weaker than covalent interactions and the interaction distances are much longer than covalent bonds and much shorter than the other non-covalent interactions. Van der Waals interactions occur at distances between 3 and 4 Å. They are very weak beyond 5Å and electron repulsion prevents atoms from getting much closer than 3Å. Van der Waals interactions are non-directional and very weak. However, significant energy of stabilization can be obtained in the central hydrophobic core of proteins by the additive effect of many such interactions.
2) hydrophobic force. The hydrophobic force is really a negative non-covalent force. The presence of hydrophobic side chains in aqueous solution induces the formation of structured water (clathrate cages of water molecule form, like miniature ice crystals about the hydrophobic side chains). This reduction in entropy of the water molecules is a very unfavorable resulting in a strong force to keep hydrophobic side chains buried in the interior of the protein. The hydrophobic force is one of the largest determinants of protein structure. Most secondary structural elements we will discuss have an amphipathic nature, one hydrophobic side and one hydrophilic side because the structure lies on the surface of the protein.
3) electrostatic forces. The attraction of oppositely charged side chains can form salt-bridges which stabilize secondary and tertiary structures. The electrostatic force is quite strong, falling off as the square of the distance between the charged atoms. It also depends heavily on the dielectric constant of the medium in which the protein is dissolved. It is strongest in a vacuum and 80 fold weaker in water, and weaker still at elevated salt solutions. Water and ions can shield electrostatic interactions reducing both their strength and distance over which they operate.
4) dipole moments. Dipole moments are caused by pairs of charges separated by a larger distance than permitting a salt- or ion bridge. The dipole moment can give rise to an electric field along the entire length of a structural element and are often used by proteins to attract and position charged substrates and products. The peptide chain naturally has a dipole moment because the N-terminus carries about 1/2 a positive charge and the C-terminus carries about 1/2 unit of negative charge. The alpha-helix is known to carry a partial negative charge at its C-terminus and an positive charge at its N-terminus. In order to help neutralize this charge distribution, alpha helices often have acidic residues near their N-terminus and a basic residue near their C-terminus.
5) hydrogen bonds. Hydrogen bonds occur when a pair of nucleophilic atoms such as oxygen and nitrogen share a hydrogen between them. The hydrogen may be covalently attached to either nucleophilic atom (the H-bond donor) and shared with the other atom (the H-bond receptor). H-bonds are very directional and their strength deteriorates dramatically as the angle changes. Hydrogen bonds do not, in general, contribute to the net stabilization energy of proteins because the same groups that hydrogen bond to each other in a native protein structure, can hydrogen bond to water in the denatured state. However, hydrogen bonds are extremely important because of their directionality, they can control and limit the geometry of the interactions between side-chains. This is shown most dramatically in patterns of hydrogen bonding between the carboxyl groups and the amino groups in the peptide backbone that give rise to alpha helix and beta strand conformations.
6) covalent bond distances and torsion angles. The major properties of the covalent bonds hold proteins together are their bond distances and bond angles. In particular, the bond angles between two adjacent bonds on either side of a single atom, or the dihedral angles between three contiguous bonds and two atoms control the geometry of the protein folding. The preferred dihedral angles for different secondary structural elements are discussed below.
Levels of Protein Structure
There are four levels of protein structure depicted below. The amino acid sequence is generally referred called the primary structure of a protein. The secondary structure is the first level of folding of the polypeptide bond and it is determined by 1) the planar nature of the peptide bond and by the phi-psi angles about the alpha carbons of each amino acid. The tertiary structure of a protein is often referred to as the fold of the protein and
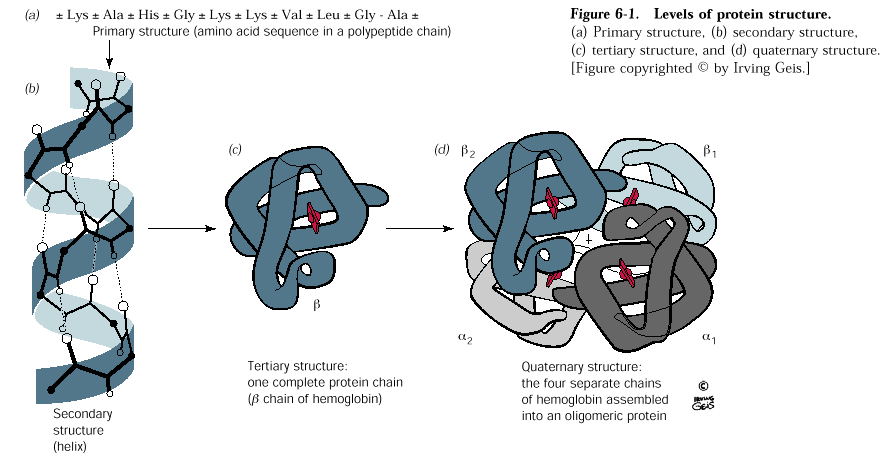
Preferred secondary structures
Alpha helices are the most well known element of protein structure, proposed by Pauling and confirmed in the first structure determined, myoglobin, alpha-helices have distinctive patterns of hydrogen bonding and phi-psi angles. They are generally between 5 and 20 residues in length, but some proteins and coiled-coil structures can be considerably longer. The carboxyl groups of the backbone hydrogen bond to the amino group of a residue four amino acids distant along the chain. Alpha helices generally have a pitch of about 3.5 residues per turn, but there are forms of helices with tighter (3 residues per turn) and longer (4 residues per turn).
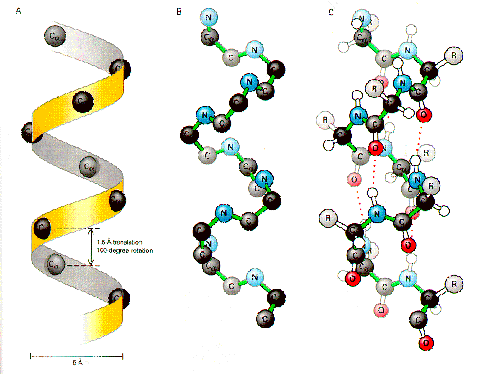
Alpha helices can be coiled about themselves in both two coil, three coil and four coil (four helix bundle) conformations. Alpha helices can exist internal in proteins (generally hydrophobic), on the surface of proteins (amphipathic) or in membranes (hydrophobic). Alpha helices can span membranes either singly or in groups.
Beta-strands are an extended form in which the side chains alternate on either side of the extended chain. The back bones of beta-strands hydrogen bond with the backbone of an adjacent beta strand to form a beta-sheet structure. The strands in a beta sheet can be either parallel or anti-parallel and the hydrogen bonding pattern is different between the two forms.
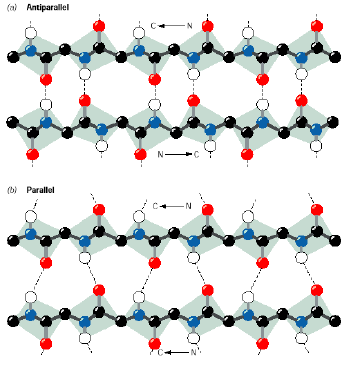
Anti-parallel beta stands are often linked by short loops containing 3-5 residues in highly characteristic conformations. Longer loops are occasionally found where the loop plays an important role in substrate binding or an active site. The antigen combining site of the immunoglobulins is an important example of this.
Beta sheets can be internal to a protein (largely hydrophobic) or on the surface in which case they are amphipathic, with every other amino acid side chain alternating between hydrophobic and hydrophilic nature.
Beta-sheets tend to be pleated or bent as shown in the figure to the left. Every other residue points to the left or to the right side of the sheet,
Rhamachandran Plots
The peptide backbone is constrained by steric hindrance, and hydrogen bonding patterns that limit its torsional angles (phi-psi angles) to certain limits. Plots of phi versus psi dihedral angles for amino acid residues are called Ramachandran plots. One can tell if the backbone is following a helical or a an extended beta strand structure based on the values of the phi-psi angles over a length of backbone (usually 3-4 residues is sufficient).
Protein Folds or Architectures

There are a limited number of ways that secondary structures interact in the tertiary structure of proteins. These architectures or folds, as they are called are cataloged in a number of protein structure databases available on the Web. The SCOP database from MRC (also maintained here at Stanford by Steven Brenner) and the CATH database from the University College of London (Janet Thornton) are two examples. The SCOP structure database is hierarchical with four subdivisions, Class (all alpha-helical, all-beta strand, alternating alpha helix and beta-strand and alpha-helix plus beta-strand), Fold, Superfamily and family. The later two classes often show sequence similarity but the first two levels of the hierarchy do not. Examples of the first three levels of the SCOP hierarchy are shown in the figure to the right.
References
Branden, C. & Tooze, J. Introduction to Protein Structure, Second Edition, Garland Publishing, New York. http://www.proteinstructure.com/
Creighton, T. E. (1993). Proteins: Structures and Molecular Properties. (Second Edition ed.). New York: Freeman.
Creighton, T. E. (Ed.). (1992). Protein Folding. New York: W. H. Freeman & Co.
Darby, N. J., & Creighton, T. E. (1993). Protein Structure. Oxford: IRL Press.
Schulz, G. E., & Schirmer, R. H. (1985). Principles of Protein Structure. New York: Springer-Verlag.
Stryer, L. (1995). Biochemistry. (Fourth Edition ed.). New York: W. H. Freeman & Co.
Internet Resources
Protein Database
http://www.rcsb.org/pdb/index.htmlSwiss-Prot Database
http://www.expasy.ch/sprot/sprot-top.htmlCATH Database of Folds
http://www.biochem.ucl.ac.uk/bsm/cath/SCOP Database
http://scop.mrc-lmb.cam.ac.uk/scop/Structural Genomics
http://structuralgenomics.org/DALI
http://www.embl-heidelberg.de/dali/dali.html3D Search
http://gene.stanford.edu/3dsearch/LOCK Protein Superposition
http://gene.stanford.edu/lock/
Mechanisms of DNA Replication
In 1971 it was known that DNA polymerases, unlike RNA polymerases, could not start new chains. In addition, it was known that there were three different DNA polymerases in Escherichia coli with different properties. The most abundant (200 molecules per cell) was DNA polymerase I. DNA polymerase II was much slower. It appeared to be primarily a repair DNA polymerase, limited to filling in short gaps resulting from repair processes. Finally, the rarer DNA polymerase III (10 molecules per cell) and was the only enzyme that could keep up with the expect rates of DNA replication fork movement (1000 nt incorporation per second).
Discovery of DNA Polymerase
The first DNA synthetic enzymes was discovered by Dr. Arthur Kornberg and his collaborators in the late 50's. They initially found that cell-free extracts from bacteria were able to incorporate
3H-thymidine into DNA much as could the intact cells, but at a very reduced rate.Two Roles for DNA in the DNA Polymerase Reaction
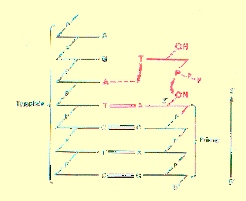
Role of DNA as Template
The DNA was found to have two roles in the synthetic reaction. The first was that of a template to direct the order of incorporation of new nucleotides. This was demonstrated several ways.
1. First the base composition of the product was the same as the template DNA.
2. Second, the frequency of dinucleotides was the same.
The Role of DNA as Primer
The second role that DNA serves in the DNA polymerase reaction is that of a primer. All of the newly synthesized DNA is covalently attached to preexisting DNA chains.
1. Proper deoxynucleoside triphosphate is chosen according to the Watson-Crick base pairing rules.
2. The 3' hydroxyl group of the primer strand, known as the primer terminus, attacks the nucleotide splitting off the terminal pyrophosphate group and the enzyme advances one base pair.
3. DNA chains only grow at the 3' end and the direction of synthesis is referred to as 5' to 3' growth. We always signify the 3' end of a DNA chain with an arrowhead.
4. The primer terminus to which the deoxynucleoside triphosphates were added had to be base paired with a single-stranded segment of DNA that was to serve as template.
5. This requirement for a free 3' hydroxyl primer terminus meant that DNA polymerase could not utilize many types of DNA normally found in nature.
RNA Polymerase Can Start Chains
One reason that the requirement for a primer terminus seemed so unusual was that all of these DNA molecules were excellent templates for the enzyme RNA polymerase. RNA polymerase is responsible for transcribing the genetic information from DNA into RNA and this enzyme can initiate RNA chains. All known DNA polymerases require a primer terminus.
Another major difficulty with this DNA synthetic mechanism is that the chains only grow in the 5' to 3' direction. No DNA polymerase has been found that can extend the 5' ends of DNA chains. This causes a problem with respect to the replication of the two daughter strands at a replication fork which of course, have opposite polarities.
How are DNA Chains Initiated?
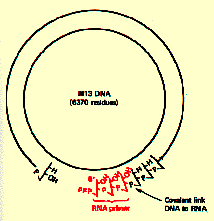 This problem was approached using the DNA from small circular single stranded phages M13, G4 and ØX174. It was known that the first step in the infection of these viral DNAs was the conversion of the viral DNA from a single stranded circular form to a duplex replicative form (RF) and this conversion was mediated by host cell enzymes alone. Hence introducing the phage DNA into bacterial extracts and isolating the factors required for the conversion to RF would reveal the mechanism of initiation of DNA chains.
This problem was approached using the DNA from small circular single stranded phages M13, G4 and ØX174. It was known that the first step in the infection of these viral DNAs was the conversion of the viral DNA from a single stranded circular form to a duplex replicative form (RF) and this conversion was mediated by host cell enzymes alone. Hence introducing the phage DNA into bacterial extracts and isolating the factors required for the conversion to RF would reveal the mechanism of initiation of DNA chains.
M13 is Primed by RNA Polymerase
Based on in vivo results showing that M13 SS ——>RF reaction was inhibited by rifampicin and other inhibitors of the host RNA polymerase, extracts of Escherichia coli were shown to convert M13 SS to a duplex form in reaction that required the four ribonucleotide triphosphates in addition to the four deoxynucleotide triphosphates. In vitro, a short RNA primer was made from several locations on the M13 circle. These primers can server to initiate DNA synthesis by DNA polymerase III.
Phages ØX174 and G4 also Primed by RNA
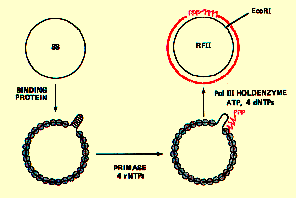 When DNA from either G4 or ØX174 is used in similar extracts, DNA synthesis is also observe. Studies with the G4 template showed that it requires only three enzymes for its SS ——> RF conversion: single-strand DNA binding protein (SSB), dnaG protein (primase) and DNA polymerase III holoenzyme (see below).
When DNA from either G4 or ØX174 is used in similar extracts, DNA synthesis is also observe. Studies with the G4 template showed that it requires only three enzymes for its SS ——> RF conversion: single-strand DNA binding protein (SSB), dnaG protein (primase) and DNA polymerase III holoenzyme (see below).
Phage ØX174 priming is much more complicated and appears to be more similar to the priming that occurs at the beginning of Okazaki fragments on the discontinuous strand of DNA synthesis at a replication fork. Initiation starts at a site known as a primer assembly site (pas) which bears no sequence similarity to the primase binding site in G4 or colE1 plasmid. There are extensive regions of DNA secondary structure however. An RNA synrhetic activity known as a primosome forms on the single-stranded phi-X174 DNA and serves to start new chains.
DNA Polymerase III Holoenzyme
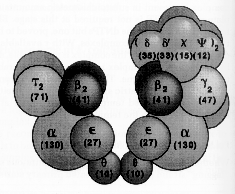
DNA polymerase III is a very complex replication protein consisting of many subunits and subassemblies. The core enzyme consists of three subunits, the
a subunit is the primary synthetic unit, the e subunit encodes the proofreading 3’ exonuclease subunit.A large form of DNA polymerase III appears to contain two copies of the
a subunit enzyme whose formation requires two b subunits are present in each half of the core dimer that give rise to extreme processivity and speed of polymerase action.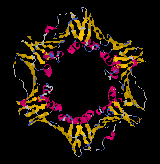
The formation of a highly processive form of the DNA polymerase III requires the binding of the
b subunits to DNA to which a core will bind. The b subunits form a rings about the DNA (like a hand cuff) to which the core DNA polymerase III associates. This hand cuff ensures that the polymerase stays locked onto the DNA and proceeds in a highly processive fashion as it synthesizes DNA. It may also permit one dimensional diffusion of the DNA polymerase III along duplex DNA once it has completed filling in a gap on the lagging strand.The Replication Fork
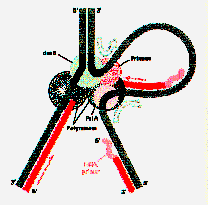 An overall model of the replication fork includes a dimer DNA polymerase III bound on the leading strand with a molecule of a DNA helicase associated with the parental DNA strand serving as template. The half of the DNA polymerase III holoenzyme with the
An overall model of the replication fork includes a dimer DNA polymerase III bound on the leading strand with a molecule of a DNA helicase associated with the parental DNA strand serving as template. The half of the DNA polymerase III holoenzyme with the
Initiation of DNA Replication
Analysis of the initiation of DNA replication at an origin was studied in much the same way as the initiation of DNA chains. A specific small molecular substrate containing an origin was designed and then used in an in vitro assay for components that would initiate replication on a duplex DNA circle
A plasmid containing the Escherichia coli origin of replication (oriC) was selected by ligating fragments of E. coli DNA with a drug resistance marker (penicillin resistance gene) and selecting penicillin resistance due to autonomously replicating plasmid circles in host E. coli cells. The size of the resulting Escherichia coli origin fragment was reduced in size by a variety of techniques until the smallest fragment that would support autonomous replication was isolated. This strategy yielded a 245 base pair minimal fragment known as the oriC fragment.
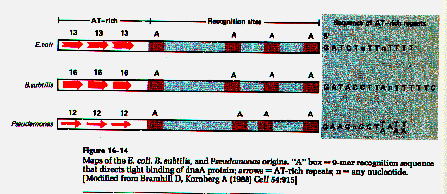
Comparison of origins from a variety of widely divergent bacterial species revealed:
1) the presence of 14 dam methylase sites supporting post-replication mismatch repair and guaranteeing accurate replication of this region. These sites also induce a high state of methylation of the origin.
2) the presence of four repeats of a 9 base sequence which is the binding site for the dnaA protein, required for chromosomal initiation.
3) the presence of a 60 base pair very AT rich region which is highly conserved in evolution.
4) the presence of three 13 base repeats in this AT rich region that bind dnaA in the prepriming reaction and melt allowing dnaB helicase to bind.
5) other regions whose sequence is less conserved but which are of fixed length in evolution
Reference
Kornberg, A. (1988). DNA Replication. J. Biol. Chem. 263 (1), 1-4.
Kuriyan, J. and O'Donnell, M. (1993). Sliding clamps of DNA polymerases. J Mol Biol 234 (4), 915-25.
RNA polymerase II transcriptional apparatus
(by Roger Kornberg)
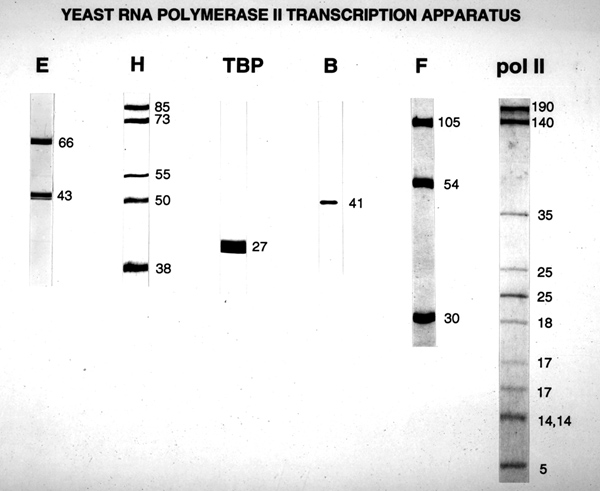
 Eukaryotic RNA polymerase II transcribes all but a few genes, RNA polymerase I transcribes the genes for the two large ribosomal RNAs, and RNA polymerase III transcribes genes for small tRNAs. RNA polymerase II from yeast has been studied in most detail. It is composed of 15 polypeptides (denoted Rpb1-12, with two copies each of Rpb3, Rpb5 and Rpb9), with a total molecular mass of 587 kDa. The two largest subunits, Rpb1 and Rpb2, exhibit extensive sequence conservation with
Eukaryotic RNA polymerase II transcribes all but a few genes, RNA polymerase I transcribes the genes for the two large ribosomal RNAs, and RNA polymerase III transcribes genes for small tRNAs. RNA polymerase II from yeast has been studied in most detail. It is composed of 15 polypeptides (denoted Rpb1-12, with two copies each of Rpb3, Rpb5 and Rpb9), with a total molecular mass of 587 kDa. The two largest subunits, Rpb1 and Rpb2, exhibit extensive sequence conservation with
Rpb1 contains an additional, unusual feature, a repeated heptapeptide at its 3'-terminus. There are 26 repeats in yeast, 45 in Drosophila, and 52 in man. The consensus heptapeptide sequence is identically conserved from yeast to man. The CTD undergoes a cycle of extensive phosphorylation and dephos-phorylation accompanying every round of transcription. The unphosphorylated form, designated IIa, initiates transcription, while the hyperphosphorylated form, designated IIo, is associated with elongation complexes.
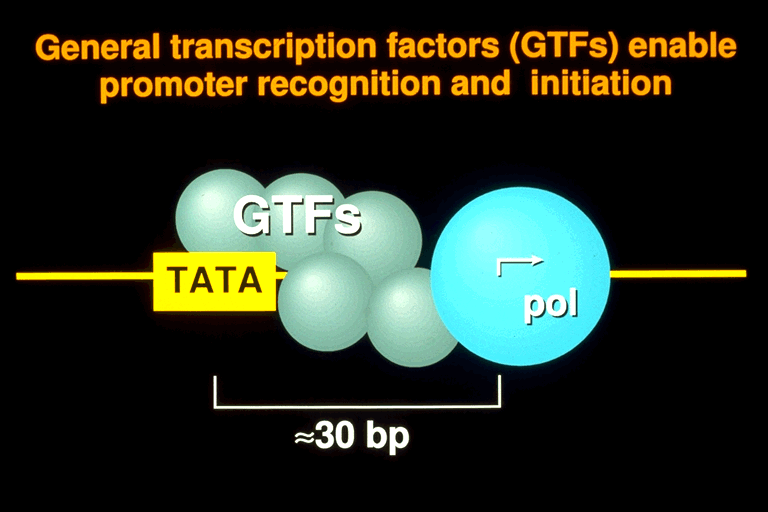 RNA polymerase II is alone incapable of recognizing a promoter and intiating transcription. It requires a set of additional proteins termed general transcription factors. Five such factors, termed TFIIB, -D, -E, -F, and -H are required for transcription of all promoters by purified RNA polymerase II in a cell-free system. TFIID can be replaced by one of its subunits, TATA-binding protein (TBP), for transcription of a core or minimal promoter, consisting of only a TATA box and transcription start site. The distance from TATA box to transcription start site is about 25 base pairs in almost all polymerase II promoters in almost all organisms. This conserved spacing is the hallmark of a polymerase II promoter.
RNA polymerase II is alone incapable of recognizing a promoter and intiating transcription. It requires a set of additional proteins termed general transcription factors. Five such factors, termed TFIIB, -D, -E, -F, and -H are required for transcription of all promoters by purified RNA polymerase II in a cell-free system. TFIID can be replaced by one of its subunits, TATA-binding protein (TBP), for transcription of a core or minimal promoter, consisting of only a TATA box and transcription start site. The distance from TATA box to transcription start site is about 25 base pairs in almost all polymerase II promoters in almost all organisms. This conserved spacing is the hallmark of a polymerase II promoter.
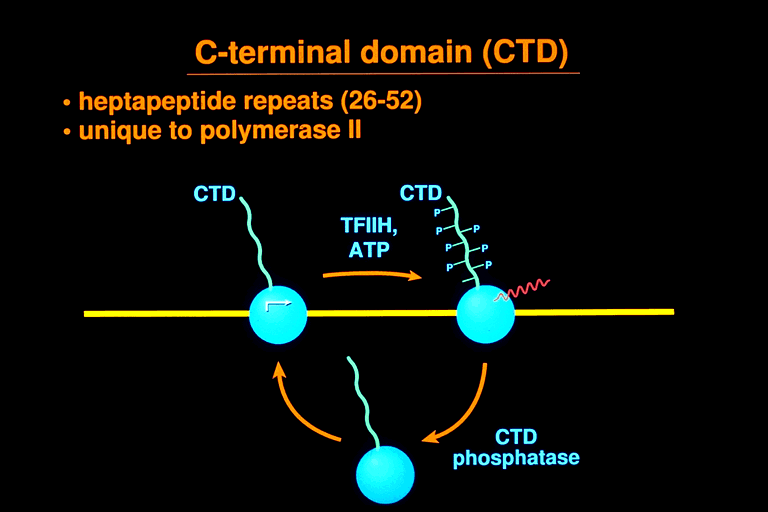 Transcription factors B, E, and F interact directly with RNA polymerase II. B couples to TBP and the TATA box, while E couples to H. The largest subunit of H is an ATPase/helicase, which unwinds double stranded DNA around the start site as required for transcription. Other subunits of H supply the kinase activity responsible for CTD phosphorylation accompanying the initiation of transcription. Both helicase and kinase subunits of H relate to other major cellular transactions. The helicase and five additional H subunits form part of a DNA repairosome, which includes other proteins required for nucleotide excision DNA repair. This connection may account for the preferential repair of DNA damage in the transcribed strand in vivo. The H kinase belongs to the family of cell cycle control protein kinases and includes a cyclin subunit as well.
Transcription factors B, E, and F interact directly with RNA polymerase II. B couples to TBP and the TATA box, while E couples to H. The largest subunit of H is an ATPase/helicase, which unwinds double stranded DNA around the start site as required for transcription. Other subunits of H supply the kinase activity responsible for CTD phosphorylation accompanying the initiation of transcription. Both helicase and kinase subunits of H relate to other major cellular transactions. The helicase and five additional H subunits form part of a DNA repairosome, which includes other proteins required for nucleotide excision DNA repair. This connection may account for the preferential repair of DNA damage in the transcribed strand in vivo. The H kinase belongs to the family of cell cycle control protein kinases and includes a cyclin subunit as well.
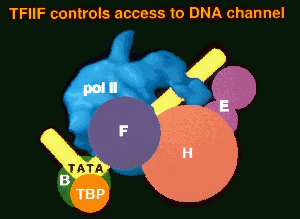 Structural studies provide a rationale for the general transcription factors and their roles in the intiation mechanism. Electron microscope crystallography reveals a cleft in E. coli RNA polymerase similar to that previously noted in the X-ray structure of the much smaller, single subunit E. coli DNA polymerase. The cleft has been likened to a hand, with palm, fingers, and thumb. Catalytic aspartate residues identify the active site in the floor (palm) of the DNA polymerase cleft. Flexibility of the thumb apparently allows entry of DNA in the cleft for initiation and assures retention in the cleft for processivity. A nearly identical tertiary fold occurs in the RNA-dependent DNA polymerase, HIV reverse transcriptase, and in the DNA-dependent RNA polymerase from bacteriophage T7.
Structural studies provide a rationale for the general transcription factors and their roles in the intiation mechanism. Electron microscope crystallography reveals a cleft in E. coli RNA polymerase similar to that previously noted in the X-ray structure of the much smaller, single subunit E. coli DNA polymerase. The cleft has been likened to a hand, with palm, fingers, and thumb. Catalytic aspartate residues identify the active site in the floor (palm) of the DNA polymerase cleft. Flexibility of the thumb apparently allows entry of DNA in the cleft for initiation and assures retention in the cleft for processivity. A nearly identical tertiary fold occurs in the RNA-dependent DNA polymerase, HIV reverse transcriptase, and in the DNA-dependent RNA polymerase from bacteriophage T7.
 Electron crystallography reveals a striking resemblence of yeast RNA polymerase II to E. coli RNA polymerase. Both enzymes exhibit multiple conformations of the arm of protein density surrounding the cleft. This arm may correspond to a zinc finger domain of E. coli
Electron crystallography reveals a striking resemblence of yeast RNA polymerase II to E. coli RNA polymerase. Both enzymes exhibit multiple conformations of the arm of protein density surrounding the cleft. This arm may correspond to a zinc finger domain of E. coli
The TATA box is recognized by TATA-binding protein, whose exceptionally conserved C-terminal 180 amino acid domain is sufficient for transcription. X-ray crystal structures have been determined for TBP alone, in a complex with TATA DNA, and in a ternary complex with TATA DNA and a large fragment of TFIIB. TBP is saddle-shaped, with a pseudo two-fold axis of symmetry relating two halves of the C-terminal domain which are similar but not identical in sequence. DNA binds to the concave underside of the saddle, at an angle to the pseudo two-fold axis. The DNA is partially unwound, from a helix to a "ladder", and TBP lies in the flat minor groove of the ladder. Kinks at the edges where DNA enters and leaves the TBP binding site result in a strongly bent shape of the bound DNA. TFIIB interacts with the DNA on both sides of TBP, whose role is, in effect, to bend DNA to enable TFIIB binding.
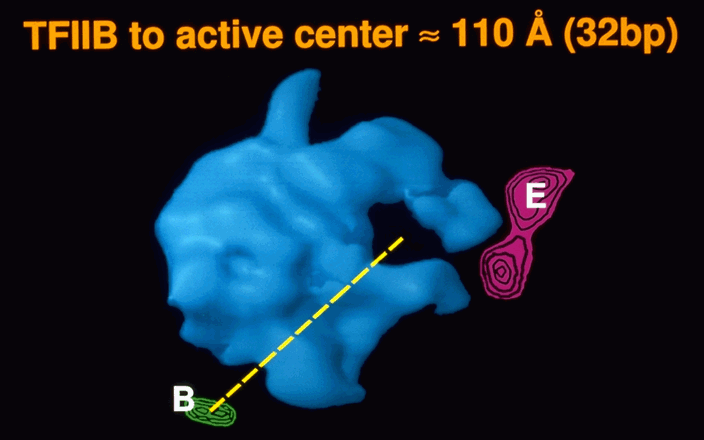
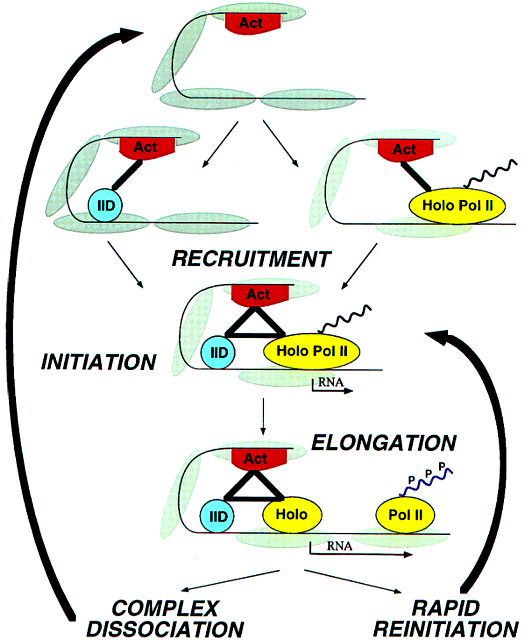
Electron crystallography of cocrystals of yeast RNA polymerase II with transcription factors B and E identifies their sites of interaction on the surface of the enzyme, leading to a simple hypothesis for the mechanism of start site determination and the intiation of transcription. B brings the TATA box of a promoter to a point on the polymerase surface about 110Å from the active center, corresponding to about 30 base pairs length of DNA, and thereby accounting for the conserved spacing of TATA box and start site of transcription. Entry of DNA triggers closure of the polymerase arm around the active center cleft which, in turn, creates the site for E binding. E recruits H, which melts the promoter to initiate transcription.
Transcriptional Activation
 Mutagenesis of RNA polymerase II promoters has identified the TATA box and upstream elements as essential for promoter activity. Upstream elements are found in the immediate vicinity of the promoter and beyond. A few main types of elements are found within the first 100 base pairs of the transcription start site of many mammalian promoters.
Mutagenesis of RNA polymerase II promoters has identified the TATA box and upstream elements as essential for promoter activity. Upstream elements are found in the immediate vicinity of the promoter and beyond. A few main types of elements are found within the first 100 base pairs of the transcription start site of many mammalian promoters.
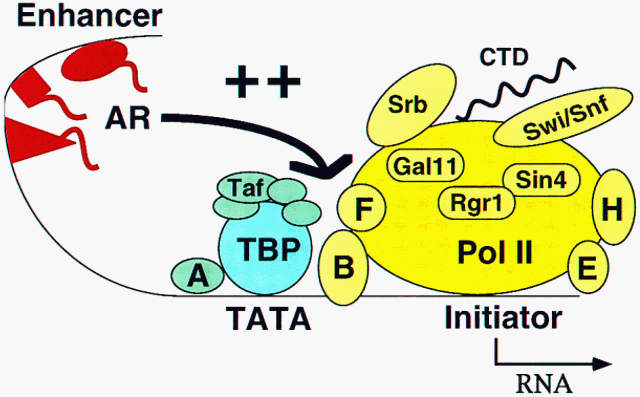 Enhancers are DNA elements distinguished by their capacity to activate transcription when located thousands of base pairs upstream of the transcription start site and when located downsteam as well. Enhancers may be responsible for regulation of transcription by inducing agents and in a temporal and tissue-specific manner. Enhancers function by interaction with activator proteins. Mechanisms of enhancer action appear to be conserved, reflecting interactions of activator proteins with components of the transcription apparatus that are homologous between yeast and man.
Enhancers are DNA elements distinguished by their capacity to activate transcription when located thousands of base pairs upstream of the transcription start site and when located downsteam as well. Enhancers may be responsible for regulation of transcription by inducing agents and in a temporal and tissue-specific manner. Enhancers function by interaction with activator proteins. Mechanisms of enhancer action appear to be conserved, reflecting interactions of activator proteins with components of the transcription apparatus that are homologous between yeast and man.
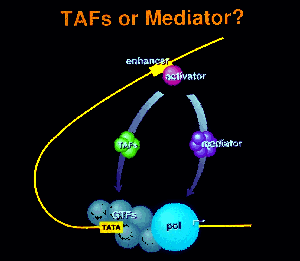
Proteins that recognize upstream promoter elements and enhancers differ from TBP in their mode of interaction with DNA. They are modular in design, with distinct, separable DNA-binding and functional ("activation") domains. The structures of many DNA-binding domains (DBDs) have been solved, and several major classes have been discerned, including helix-turn-helix (HTH), zinc finger, and leucine zipper or bHLH-ZIP families of DBDs.
 Activator proteins communicate with the basal transcription machinery at promoters through intermediary factors. Two candidates for such factors have been identified, the TAF complex which interacts with TBP, and the Mediator complex which interacts with the RNA polymerase II CTD. TAFs enable a response to activators in a partially reconstituted Drosophila or human transcription system, while Mediator supports activation in a fully defined yeast system. Deletion or destruction of TAFs has, however, no effect upon induction or transcription of most genes in yeast in vivo. Inactivation of Mediator components, on the other hand, abolishes both induction of specific genes and transcription in general. It therefore appears that Mediator is the primary conduit of information from enhancers to promoters in vivo.
Activator proteins communicate with the basal transcription machinery at promoters through intermediary factors. Two candidates for such factors have been identified, the TAF complex which interacts with TBP, and the Mediator complex which interacts with the RNA polymerase II CTD. TAFs enable a response to activators in a partially reconstituted Drosophila or human transcription system, while Mediator supports activation in a fully defined yeast system. Deletion or destruction of TAFs has, however, no effect upon induction or transcription of most genes in yeast in vivo. Inactivation of Mediator components, on the other hand, abolishes both induction of specific genes and transcription in general. It therefore appears that Mediator is the primary conduit of information from enhancers to promoters in vivo.
A small number of yeast promoters do require TAFs for transcription in vivo, including those for G1 cyclins and for some cell cycle-independent genes. Dissection of these promoters identifies sequences surrounding the TATA box and not upstream activating elements as responsible for the TAF requirement. TAFs are evidently involved in promoter selection and specificity rather than enhancer-promoter interaction.