| Bioinformatik: Einführung und Arbeiten an der GMD Thomas Lengauer Einführung |
Die Bioinformatik führt die beiden heute im Zentrum des technischen Fortschritts stehenden Disziplinen Informationstechnik und Molekularbiologie zusammen. Sie ist ein anwendungsorientierter Zweig der Informationstechnik und führt zu Schlüsselprodukten, meistens Softwaresystemen, die bei der auf molekularbiologischen Wissen basierenden Diagnose und Therapie von Krankheiten sowie beim Medikamentenentwurf eingesetzt werden.
In den letzten zehn Jahren wurde damit begonnen, systematisch die gesamte Erbinformation ganzer Organismen experimentell zu ermitteln. Der Bauplan eines jeden Lebewesens ist als Genom in der DNS (Desoxyribonukleinsäure) jeder Zelle des Organismus codiert, und zwar als lineare Abfolge kleiner molekularer Baueinheiten, die sogenannten Basen die mit den Buchstaben A, C, G und T (für die vier Nukleotide Adenin, Cytosin, Guanin und Thymin) bezeichnet werden. Da DNS im allgemeinen als Doppelhelix vorliegt, in der jeweils zwei komplement"are Basen gepaart sind, gibt man die Größe von Genomen meist in Basenpaaren (bp) an. Sie reicht von wenigen tausend bei einigen Viren über einigen Millionen bei Bakterien bis zu vielen Milliarden bei hoch entwickelten Lebewesen. Das Genom des Menschen hat etwa 3 Milliarden Basenpaare. Die Aufklärung des gesamten Genoms eines Organismus stellt den vollständigen Bauplan für das Lebewesen zur Verfügung. Derzeit sind über ein Dutzend Genome von medizinisch oder biotechnologisch wichtigen Bakterien vollständig ermittelt, darüber hinaus das Genom des Eukaryonten Hefe, dessen Zellorganisation bereits der höher entwickelter Lebewesen ähnelt. Wichtige Bestandteile des Genoms sind die Gene. Das sind sogenannte codierende Abschnitte des Genoms, die den Bauplan der vom Organismus benötigten Proteine darstellen. Daneben hat das Genom auch sogenannte nichtcodierende Regionen, die unter anderem die Übersetzung der Gene steuern, deren Rolle aber bis heute weniger gut verstanden ist.
Die Verfügbarkeit des Genoms eines Organismus bildet die prinzipielle Grundlage für ein vollständiges Verstehen der molekularen Bestandteile und Prozesse dieses Lebewesens. Dazu muß man jedoch im einzelnen aufklären, wie der Organismus die genomische Information interpretiert. Diese Aufgabe kann als die größte Herausforderung der Molekularbiologie für die kommenden Generationen angesehen werden. Einige Aspekte sind hier bereits geklärt. So kennt man den genetischen Code , mit dem die Basenpaare eines Gens in die Grundbausteine der Proteine (20 Aminosäuren) übersetzt werden. Dieser Code ist im wesentlichen bei allen Lebewesen derselbe. Darüber hinaus wird die Interpretation genomischer Information jedoch schnell sehr schwierig.
(a)
(b)
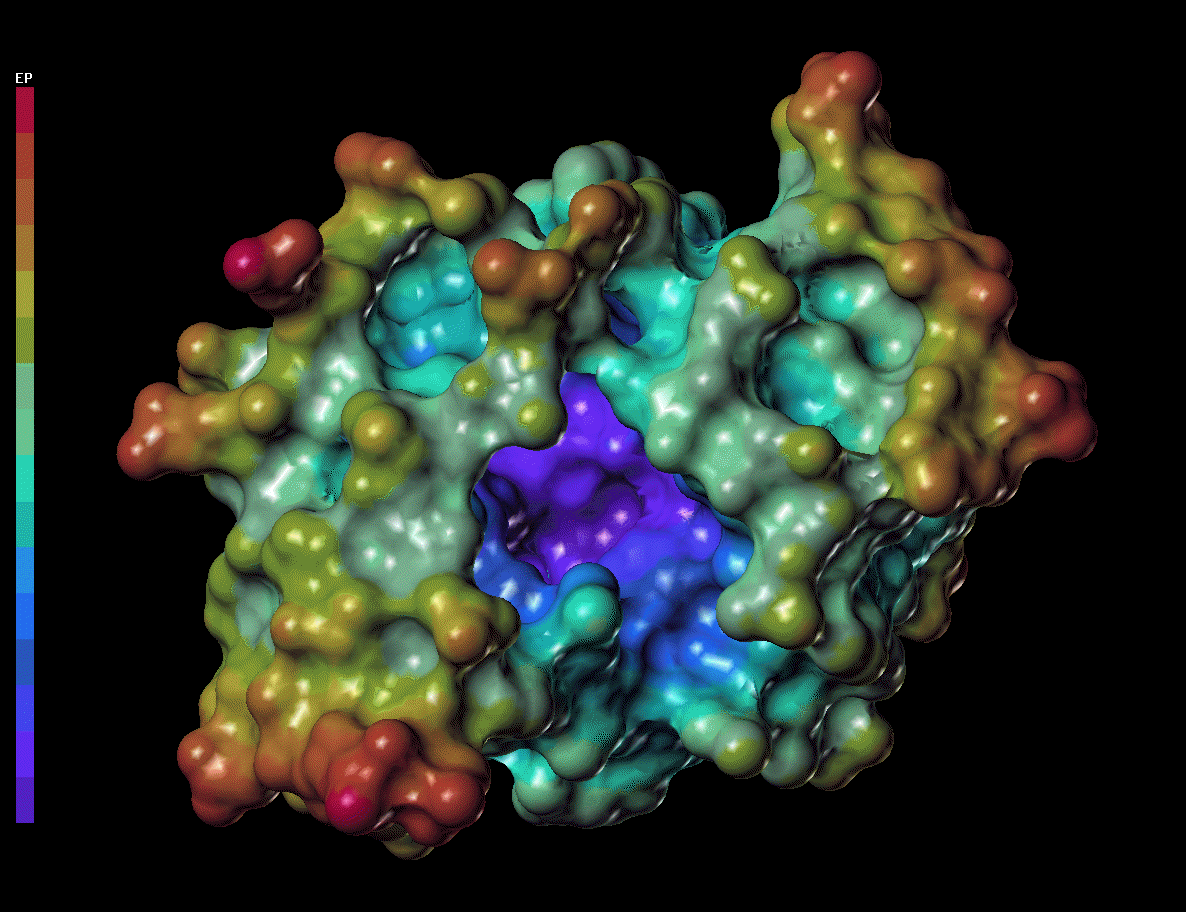
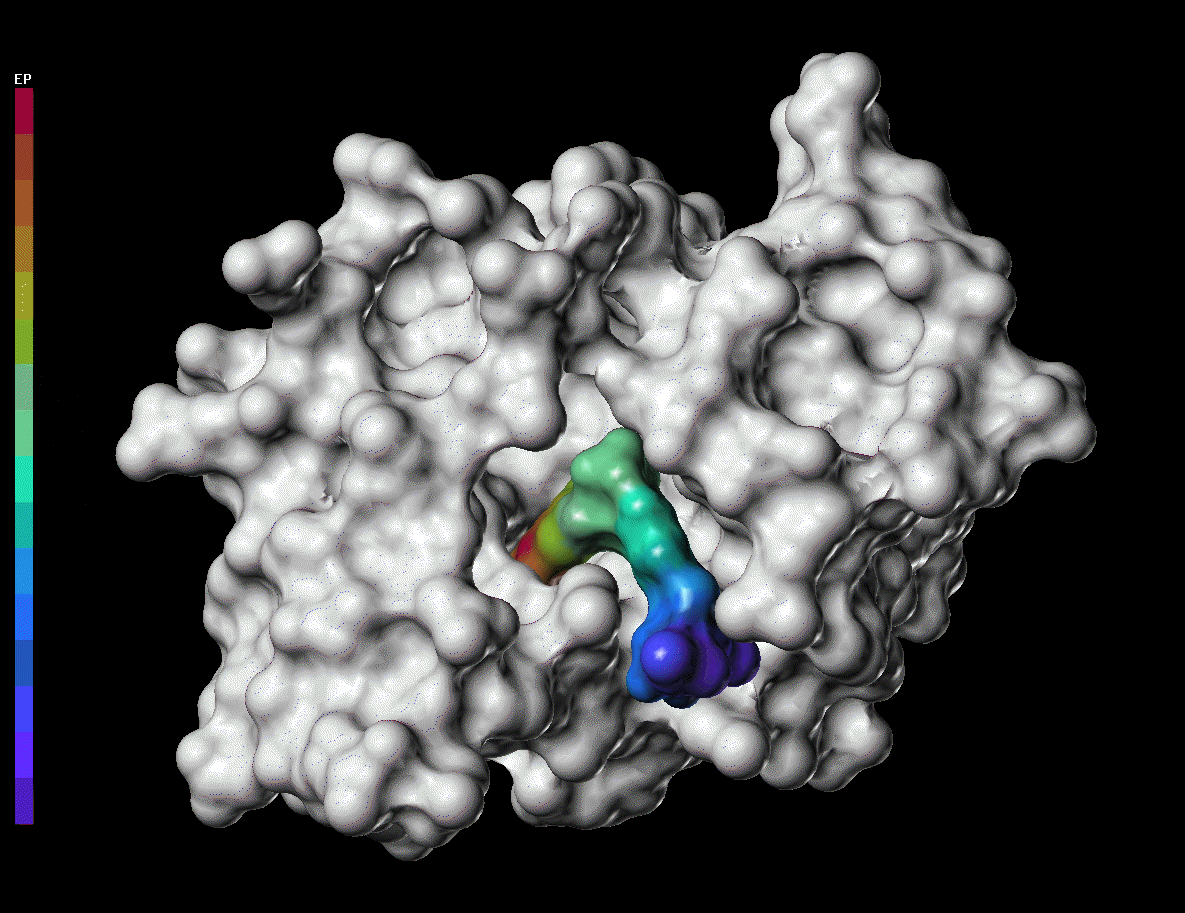
Abbildung 1 Ein Protein ohne (a) und mit (b) einem daran bindenden Wirkstoffmolekül. Die in (a) deutlich erkennbare Grube im Protein dient dazu, ein bestimmtes im Körper vorhandenes Molekül zu binden und zu verändern. Durch die Bindung des Wirkstoffmoleküls (b) wird diese Aktivität des Proteins blockiert. Die Farben stellen die elektrische Ladung an der Proteinoberfläche dar.
Wie die DNS sind auch Proteine lange molekulare Ketten, die aus bis zu mehreren hundert Aminosäuren bestehen. Diese Ketten falten sich spontan oder im Beisein von Helferproteinen in einer eindeutigen Art und Weise (siehe Abbildung 1). Die dadurch erhaltene Raumstruktur des Proteins bildet die Grundlage für seine Funktion. Proteine sind zum Teil Bausteine für den Körper, zum Teil aber auch molekulare Maschinen, die an andere Moleküle binden und so Stoffe verändern oder transportieren bzw. Signale weiterleiten. So bindet das Protein aus Abbildung 1 an der deutlich erkennbaren Grube, dem sogenannten aktiven Zentrum, zwei kleine organische Moleküle und verändert eines davon. Auf diese Weise können die Proteine als die "Zahnräder" des Mechanismus Organismus verstanden werden. Ihr Aussehen und die Art und Weise, wie sie an andere Moleküle binden, bestimmen die molekularbiologischen Prozesse, die im Organismus vor sich gehen. Abweichung in Aussehen und Funktion dieser Moleküle sind die Grundlage für Krankheiten. Die Beeinflussung ihrer Funktion, etwa durch Hemmung, d.h. Blockieren des aktiven Zentrums mit einem spezifisch dafür geformten Wirkstoffmolekül ist die zentrale Absicht beim Medikamentenentwurf.
Aufgaben der Bioinformatik
Durch die Verfügbarkeit genomischer Information wird die Biologie von einer rein phänomenologischen beschreibenden Wissenschaft zu einer analytischen erklärenden Disziplin. Dieser Paradigmenwechsel ist mit dem verglichen worden, den die Chemie vor etwa hundert Jahren erfuhr, als das Periodensystem der Elemente die scheinbar unbegrenzte Stoffvielfalt auf eine endliche Menge von chemischen Elementen reduzierte. Nur ist der Datenbestand in der Biologie ungleich höher, insbesondere, wenn man berücksichtigt, daß die Genome etwa zweier Menschen sich, wenn auch sehr wenig, so doch an entscheidenden Stellen unterscheiden.
Der Bioinformatik kommen in diesem Umfeld zwei Aufgaben zu. Zum einen begleitet sie den Prozeß der Sequenzierung, also das mit neuartigen Laborautomaten durchgeführte Ablesen des Genoms. Hier werden heute bis zu über einer Million Basenpaare pro Tag verarbeitet. Zum anderen stellt sie komplexe Methoden der Datenanalyse zur Verfügung, um den zunächst unverständlichen genomischen Text zu interpretieren, z.B. die Stellen zu finden, wo Gene liegen, mögliche dreidimensionale Strukturen der entsprechenden Proteine vorzuschlagen und die Funktionen von Proteinen mit den ihnen zugeordneten Partnermolekülen, den sogenannten Liganden zu untersuchen.
Vor allem in den Bereichen, die Raumstruktur und Funktion von Molekülen betreffen, hängt man weiterhin entscheidend vom Laborexperiment ab, aber die verfügbare Datenmenge ist so groß, daß man sie im Labor auch nicht annähernd vollständig untersuchen kann. Eine Analyse großer Mengen verschiedener Moleküle zur Findung von guten Kandidaten für einen bestimmen Zweck nennt man Screening. So screent man etwa die Gene eines gesamten Organismus (unter Einsatz von speziell dafür entwickelten DNS Chips), um diejenigen Proteine zu finden, die für eine bestimmte gesunde oder krankhafte Lebensfunktion entscheidend sind. Oder man screent große Mengen potentieller Wirkstoffmoleküle, um ihre Qualität als Hemmstoffe für ein Protein zu ermitteln. Screening hat immer eine Komponente, die Daten im Rechner analysiert, um die guten Kandidaten zu ermitteln. Screening kann dabei aber auch vollständig im Rechner ablaufen, wenn die verwendeten Molekülmodelle hinreichend genau sind.
Produkte der Bioinformatik
Die Produkte der Bioinformatik sind also unter anderem Programme, mit denen genomische Information gescreent werden kann. An der GMD konzentrieren wir uns dabei auf zwei Themengebiete. Zum einen haben wir mit dem System ToPLign (Toolbox for Protein Alignment) ein Programmsystem entwickelt, das die Raumstruktur von Proteinen vorhersagt. Wir tun dies wiederum durch Screening. Diesmal wird die molekulare Kette des zu analysierenden Proteins, gegen eine Datenbank von einigen hundert bis einigen tausend strukturbekannten Proteinen gescreent, um das Protein zu ermitteln, dessen Struktur der des zu analysierenden Proteins wahrscheinlich am meisten ähnelt. Diese Auswahl führt das Programm in über der Hälfte der Fälle korrekt durch, was für heutige Verhältnisse beachtlich ist. In diese Software arbeiten wir zur Zeit Methoden ein, um die mit DNS Chips ermittelten Labordaten zu analysieren und so Proteine zu ermitteln, die für Krankheitsgeschehen zentral sind und damit den Ausgangspunkt für neuartige Therapien darstellen können.
Unser zweiter Forschungsbereich konzentriert sich auf molekulares Docking, das heißt auf die Untersuchung der Wechselwirkungen zwischen Proteinen und potentiellen Wirkstoffmolekülen. Hier haben wir das Dockingprogramm FlexX entwickelt, das in etwa einer Minute die Struktur des molekularen Komplexes zwischen einem, in seiner Struktur bekannten, Protein und einem Wirkstoffmolekül (Ligand) berechnet und die entsprechende freie Bindungsenergie abschätzt. Den letzteren Wert braucht man, um die Wirksamkeit eines Medikamentes zu beurteilen. FlexX ist weltweit das schnellste Programm seiner Art und macht in etwa 70% aller Fälle richtige Vorhersagen. Es ist das einzige Programm, mit dem heute das Screenen ganzer Datenbanken von Wirkstoffmolekülen möglich ist. Eine Variante FlexS, die ebenfalls bei uns entwickelt wurde, zielt darauf ab, aktive von inaktiven Substanzen zu unterscheiden, wenn die Struktur des Proteins nicht bekannt ist. In diesem Fall werden als wirksame Medikamente bekannte Stoffe mit dem Kandidatenmolekül verglichen.
Methoden der Bioinformatik
In der Bioinformatik kommen eine Vielzahl von Informatikmethoden zum Einsatz. An der GMD konzentrieren wir uns besonders auf algorithmische Methoden, etwa Methoden der dynamischen Programmierung und kombinatorischen Optimierung zur Ähnlichkeitsanalyse molekularer Sequenzen, Methoden der algorithmischen Geometrie zur Strukturanalyse von Molekülen sowie statistische Methoden des Pattern Matching und maschinellen Lernens zur Kalibrierung von Programmen zur Proteinstrukturvorhersage. Dabei kommt der Fall, daß bereits ausentwickelte Informatikmethoden einfach in die Bioinformatik übernommen werden können, praktisch nicht vor. Das liegt zum einen daran, daß die Problemstellungen in der Bioinformatik nicht in dem Sinne generisch sind, daß sie im anwendungsfreien Raum in natürlicher Weise auftreten. Vielmehr werden sie durch zusätzliche biologische und chemische Randbedingungen dominiert. Ferner richten sich auch die Bewertungskriterien für die Güte von Algorithmen nach der biologischen Relevanz der Resultate und unterscheiden sich damit von klassischen Bewertungsstandards der Informatik.
Weitere für die Bioinformatik zentrale Methoden sind die Computergraphik, hier ist jedoch bereits viel an Entwicklung geschehen, und Datenbankmethoden.
| [ Vorlesung | Praktikum | Zeitplan der Vorlesung | neue Praktikumsinfo | Literatur | Anfang ] |